Overview
R-itable estimates narrow-sense heritability (h²) for quantitative traits in family cohort studies using a profile-likelihood variance-components approach, without requiring SOLAR Eclipse or any proprietary software.
The three main steps are:
- Build the additive genetic relationship matrix (GRM) from a pedigree.
- Estimate heritability for one trait with
herit_vc(). - Scale to many traits with
herit_batch(), then visualise withplot_forest().
1. Build the GRM
build_grm() takes a pedigree data frame and returns an
additive genetic relationship matrix (GRM, also written
A). The GRM encodes how much genetic material any two
individuals share due to common ancestry, and is the cornerstone of the
variance-components model.
The pedigree needs four columns:
| Column | Content |
|---|---|
id |
Individual ID |
pat |
Father’s ID (0 or NA for founders) |
mom |
Mother’s ID (0 or NA for founders) |
sex |
1 = male, 2 = female |
Importantly, the pedigree should contain everyone —
both the study subjects you want to analyse and their
parents/grandparents (founders), even if the founders have no phenotype
data. Including founders lets kinship2 trace shared
ancestry correctly.
In the example below, IDs 1–4 are two unrelated founder couples. IDs 5–6 are offspring of couple 1×2, and IDs 7–8 are offspring of couple 3×4. The study subjects are IDs 5–8.
ped <- data.frame(
id = 1:8,
pat = c(0, 0, 0, 0, 1, 1, 3, 3), # founders have pat = 0
mom = c(0, 0, 0, 0, 2, 2, 4, 4), # founders have mom = 0
sex = c(1, 2, 1, 2, 1, 2, 1, 2)
)
A <- build_grm(ped, study_ids = 5:8)
round(A, 3)
#> 5 6 7 8
#> 5 1.0 0.5 0.0 0.0
#> 6 0.5 1.0 0.0 0.0
#> 7 0.0 0.0 1.0 0.5
#> 8 0.0 0.0 0.5 1.0Reading the matrix:
- Diagonal = 1 for non-inbred individuals (each person shares 100% of their genome with themselves).
- 5 & 6 share 0.5 — they are full siblings (same parents 1×2), so they share on average half their segregating alleles.
- 7 & 8 share 0.5 — full siblings of couple 3×4, same logic.
- 5 & 7 share 0 — their parents come from entirely different founders, so they are genetically unrelated in this pedigree.
This block-diagonal pattern is typical of a study with multiple independent families. In a real cohort the pedigree is often more complex (multi-generation, distant relatives), but the interpretation is the same: larger off-diagonal values mean more shared ancestry.
2. Estimate heritability for a single trait
set.seed(42)
# Simulate some phenotype data for the study subjects
dat <- data.frame(
IID = 5:8,
age = c(35, 38, 40, 44),
sex_num = c(1, 2, 1, 2),
bmi = c(24.1, 27.3, 22.8, 29.5)
)
# Unadjusted model
res_unadj <- herit_vc("bmi", grm = A, data = dat, min_n = 3)
#> ✔ bmi_unadj n=4 h2=0.001 [0,1] p=0.5
# Adjusted model (age + sex)
res_adj <- herit_vc("bmi", grm = A, data = dat,
covs = c("age", "sex_num"),
label = "bmi_adj",
min_n = 3)
#> ✔ bmi_adj n=4 h2=0.001 [NA,NA] p=0.5
str(res_unadj)
#> List of 12
#> $ label : chr "bmi_unadj"
#> $ trait : chr "bmi"
#> $ covariates : chr ""
#> $ n : int 4
#> $ h2 : num 0.001
#> $ se : num 44.7
#> $ ci_lo : num 0
#> $ ci_hi : num 1
#> $ pval : num 0.5
#> $ var_covariates: num NA
#> $ sigma2_a : num 0.00071
#> $ sigma2_e : num 0.712The returned list contains:
| Field | Description |
|---|---|
h2 |
MLE narrow-sense heritability |
se |
SE from profile-LL curvature |
ci_lo, ci_hi
|
95% profile-likelihood CI |
pval |
One-sided LRT p-value (boundary-corrected) |
var_covariates |
Variance explained by fixed-effect covariates (R²); NA
for unadjusted models |
sigma2_a |
Additive genetic variance (σ²_g) |
sigma2_e |
Residual environmental variance (σ²_e) |
3. Batch estimation
For real analyses you will typically run many traits across several
covariate models. herit_batch() handles this and returns a
tidy data frame.
# Add a second trait
dat$hdl <- c(55.0, 60.2, 48.3, 72.1)
covs_list <- list(
unadj = NULL,
cov1 = c("age", "sex_num")
)
res <- herit_batch(
traits = c("bmi", "hdl"),
grm = A,
data = dat,
covs_list = covs_list,
min_n = 3,
.progress = FALSE
)
res
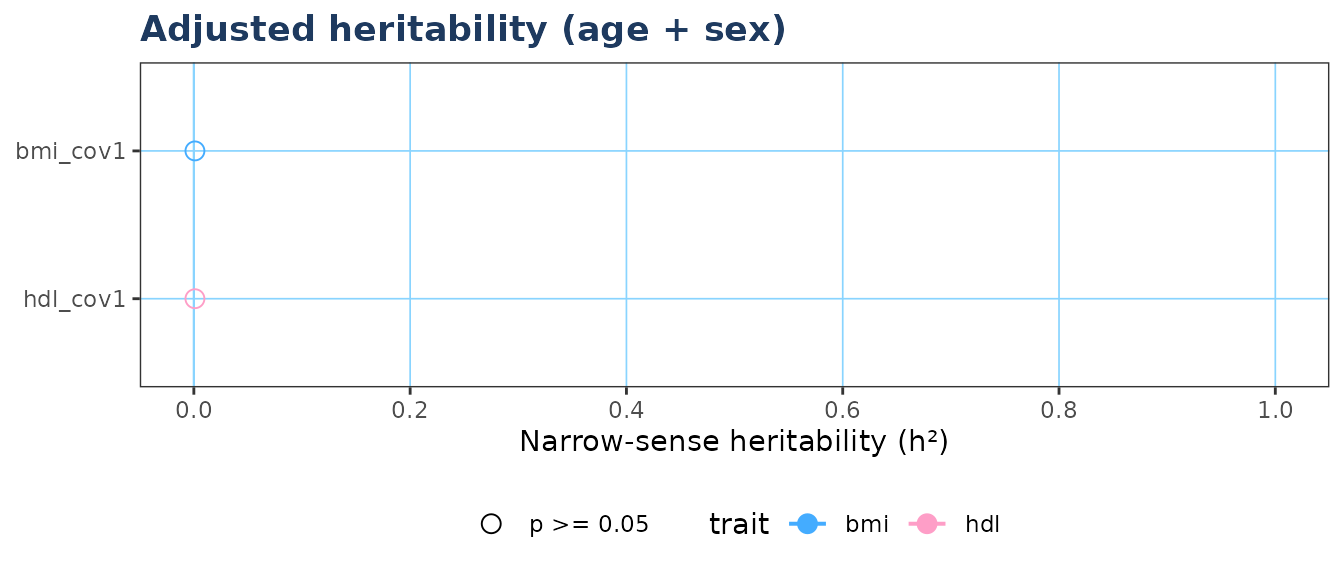
#> label trait covariates n h2 se ci_lo ci_hi pval var_covariates
#> 1 bmi_unadj bmi 4 0.001 44.7214 0 1 0.5 NA
#> 2 hdl_unadj hdl 4 0.001 44.7214 0 1 0.5 NA
#> 3 bmi_cov1 bmi age+sex_num 4 0.001 NA NA NA 0.5 0.7592
#> 4 hdl_cov1 hdl age+sex_num 4 0.001 NA NA NA 0.5 0.7592
#> sigma2_a sigma2_e
#> 1 0.00071 0.71206
#> 2 0.00071 0.71206
#> 3 0.00017 0.17143
#> 4 0.00017 0.171434. Forest plot
if (requireNamespace("ggplot2", quietly = TRUE)) {
plot_forest(res,
model_filter = "cov1",
colour_by = "trait",
title = "Adjusted heritability (age + sex)")
}
5. Colour palette
Ritable_colours is exported for consistent figure
styling across your analysis. It contains the four brand colours used in
plot_forest() by default:
Ritable_colours
#> pink blue sky cream
#> "#FE9EC7" "#44ACFF" "#89D4FF" "#F9F6C4"You can use them directly in ggplot2 calls:
ggplot2::scale_colour_manual(values = Ritable_colours[c("blue", "pink")])6. Realistic workflow
Here we simulate a small multi-family cohort — 80 nuclear families, 2 parents + 3 offspring each — with three phenotypes that have different true h² values. This mirrors what you would do with real pedigree and phenotype CSV files.
library(Ritable)
# ---- 1. Simulate pedigree ---------------------------------------------------
set.seed(2026)
n_fam <- 80
ped <- do.call(rbind, lapply(seq_len(n_fam), function(f) {
base <- (f - 1L) * 5L
data.frame(
id = base + 1:5,
pat = c(0L, 0L, base + 1L, base + 1L, base + 1L),
mom = c(0L, 0L, base + 2L, base + 2L, base + 2L),
sex = c(1L, 2L, sample(1:2, 3, replace = TRUE))
)
}))
# ---- 2. Simulate phenotypes for offspring -----------------------------------
# Three traits with approximate h2: bmi ~0.6, sbp ~0.35, hdl ~0.5
simulate_trait <- function(ped, n_fam, h2, seed) {
set.seed(seed)
genetic <- rep(rnorm(n_fam, 0, sqrt(h2)), each = 5)
residual <- rnorm(nrow(ped), 0, sqrt(1 - h2))
genetic + residual
}
pheno_all <- data.frame(
bmi = simulate_trait(ped, n_fam, h2 = 0.60, seed = 1),
sbp = simulate_trait(ped, n_fam, h2 = 0.35, seed = 2),
hdl = simulate_trait(ped, n_fam, h2 = 0.50, seed = 3)
)
off_rows <- ped$pat != 0L
dat <- data.frame(
IID = ped$id[off_rows],
age = round(runif(sum(off_rows), 20, 75)),
sex_num = ped$sex[off_rows],
bmi = pheno_all$bmi[off_rows],
sbp = pheno_all$sbp[off_rows],
hdl = pheno_all$hdl[off_rows]
)
dat$age2 <- dat$age^2
# ---- 3. Build GRM once — reuse for all models -------------------------------
A <- build_grm(ped, study_ids = dat$IID)
# ---- 4. Define covariate models ---------------------------------------------
covs_list <- list(
unadj = NULL,
cov1 = c("age", "sex_num"),
cov2 = c("age", "sex_num", "age2")
)
# ---- 5. Batch estimation ----------------------------------------------------
res <- herit_batch(
traits = c("bmi", "sbp", "hdl"),
grm = A,
data = dat,
covs_list = covs_list
)
res
#> label trait covariates n h2 se ci_lo ci_hi pval
#> 1 bmi_unadj bmi 240 0.9990 0.1297 0.8400 1 0
#> 2 sbp_unadj sbp 240 0.8055 0.1392 0.5303 1 0
#> 3 hdl_unadj hdl 240 0.7998 0.1394 0.5243 1 0
#> 4 bmi_cov1 bmi age+sex_num 240 0.9990 0.1313 0.8331 1 0
#> 5 sbp_cov1 sbp age+sex_num 240 0.8012 0.1402 0.5240 1 0
#> 6 hdl_cov1 hdl age+sex_num 240 0.8014 0.1395 0.5258 1 0
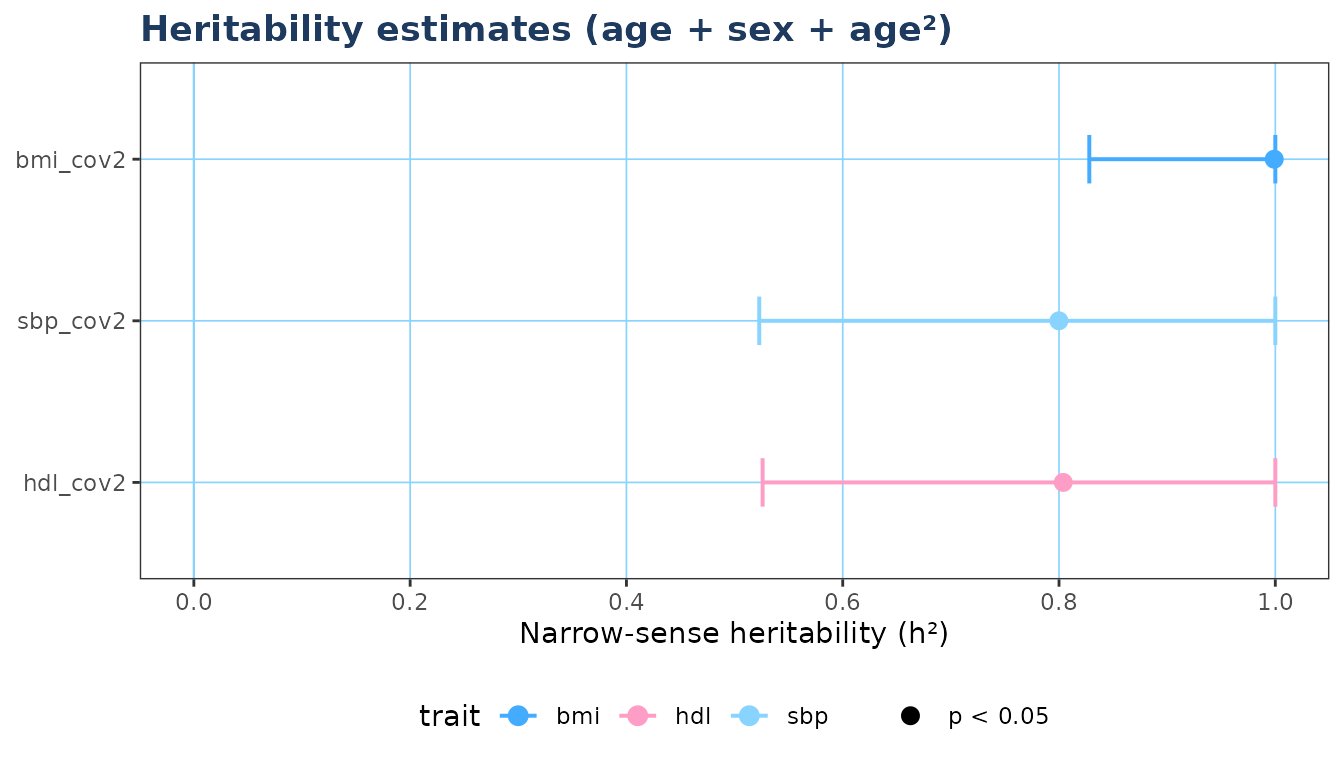
#> 7 bmi_cov2 bmi age+sex_num+age2 240 0.9990 0.1318 0.8280 1 0
#> 8 sbp_cov2 sbp age+sex_num+age2 240 0.7999 0.1402 0.5228 1 0
#> 9 hdl_cov2 hdl age+sex_num+age2 240 0.8039 0.1405 0.5259 1 0
#> var_covariates sigma2_a sigma2_e
#> 1 NA 0.93477 0.00094
#> 2 NA 0.80117 0.19348
#> 3 NA 0.79550 0.19915
#> 4 0.0207 0.92785 0.00093
#> 5 0.0085 0.79187 0.19651
#> 6 0.0067 0.79210 0.19624
#> 7 0.0304 0.92432 0.00093
#> 8 0.0118 0.78783 0.19710
#> 9 0.0091 0.79525 0.19393
if (requireNamespace("ggplot2", quietly = TRUE)) {
plot_forest(res, model_filter = "cov2",
title = "Heritability estimates (age + sex + age\u00b2)")
}